bmjhayward blog and writings
I write about science and technology, mostly. I live in Newcastle Australia and try to enjoy the good moments.
Some applications of Alphafold 2 The AI is useful beyond general protein structure predictions.
Alphafold has successfully predicted the structure of over 98% of the human genome6. The annual “Critical Assessment of protein Structure Prediction” CASP is an annual world wide experiment where research groups develop models and apply them to sequence data made available by UC Davis. The results in CASP14 had a significant impact on current and future possible research. Now with CASP15 happening throughout this year, it will be very interesting to see the results come December.
A brief introduction to AlphaFold and Neural Networks
If you’re not big into machine learning, but would like to know the “gist” of how AF2 works, here it is:
- Given a sequence, it produces a multi-sequence alignment (MSA) and pairwise distance plots.
- These are fed into the “Evoformer” neural network to create single and pairwise residue heatmaps.
- These outputs are used to make predictions which can feed back into Evoformer, improving future results.
If you’re interested in neural networks but don’t have time for all the theory, here’s the gist on those as well:
- Imagine a spread-sheet-like structure with functions and macros on every column.
- Create more spread sheets like the above if you like, with their own functions and macros.
- Link them together so when one completes its calculations it passes them on to the next.
- Pass your data into the first spreadsheet, read your results from the last.
- Profit! 5a. There is often a step called “backpropagation” where the neural network will pass the results back up the chain of spreadsheets, correcting and tweaking parameters in each function to optimise results.
For more details, I highly recommend this blog post from the Oxford Protein Informatics Group: https://www.blopig.com/blog/2021/07/alphafold-2-is-here-whats-behind-the-structure-prediction-miracle/.
For still more details, the blog post from DeepMind: https://www.deepmind.com/blog/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology. If you’re still hungry for knowledge, here is the paper from DeepMind, published in Nature: https://www.nature.com/articles/s41586-021-03819-2
While intended for single chain targets, it was shown that AlphaFold can also accurately model homo-oligomers. Since CASP14, the team at DeepMind have developed AlphaFold-Multimer and AlphaDesign. These tools enable prediction on homo- or hetero-oligomers and protein design, respectively.
While you may well be aware of its ability to predict novel protein structure, AF has several other use cases as well:
- starting models for electron microscopy and crystallography
- investigate IDPs; intrinsically disordered proteins that don’t keep their shape on their own
- guiding synthesis/construct design of proteins
- predict alternative conformations
- predict effects of mutation
This article is an attempt to skim across the top of research and development using AlphaFold and briefly summarise what some clever folks have been up to.
Acronyms, terms and technology used in this article: AF: alphafold AF2 alphafold 2 AFM: alphafold multimer AFD: alphafold design
RoseTTa, or “rosettaFold” is protein structure prediction technology with a different approach to AF. It’s used in several of the examples here and is widely used. Rosetta and AlphaFold seem to be used in a complementary fashion in many cases which is an interesting trend.
SEQ, Seq or seq. Short for “sequencing”. E.g. RNA-seq. The technique/practice of sequencing DNA/RNA/proteins. Very common and is used in a lot of acronyms, shorthand, technology names and brand names.
AlphaFold accurately predicts distinct conformations based on oligomeric state of a de novo designed protein1
It had been hypothesised AF could be useful in the design of protein switches. For example, a “switch” to turn hunger on or off, or to interrupt allergic reaction. To investigate, they designed a protein using RoseTTA, named SEWN0.1. It binds with “G-alpha-q” (Gαq hereafter) . Gαq is an oncogene implicated in many types of melanoma. The authors were looking for competitive inhibitors as a potential treatment. To put it another way, if they could find or create something that binds with Gαq, it could be used to block or silence the gene, and prevent development of new skin cancers.
In the general sense, this kind of “protein switching” would have almost limitless medical potential and would illuminate many, many biological processes.
RosettaFold has an optional protocol called “SEWING” which takes substructures from proteins and recombines them to make new ones. It uses geometric algorithms to compare the substructures and rearranges then into new backbones.
Using this SEWING protocol they replicated one of the helix-turn-helix substructures from a protein named PLC-beta-3 (PDB 3OHM).
SEWN0.1 is a new sequence and doesn’t have any homologs in nature as far as the authors are aware. It was designed before AF existed and wasn’t included in any training data.
AF2 then accurately predicted its structure, including the Gαq binding site! Even though it had never seen a structure like it before!
They used the AlphaFold2.ipynb v1.0 colab notebook which is part of the ColabFold framework. You can try it out for yourself here: https://github.com/sokrypton/ColabFold
 Superpositions of RoseTTaFold models on the predicted AlphaFold structures. Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/02/03/2022.02.02.478886/F2.large.jpg
Superpositions of RoseTTaFold models on the predicted AlphaFold structures. Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/02/03/2022.02.02.478886/F2.large.jpg
Predicting the structure of large protein complexes using AlphaFold and sequential assembly2
For this study the authors used a fairly common desktop computer with 2 graphics cards installed to run AF and AFM. On this platform the authors found about 3000 residues were the limit of what this approach could compute. So anyone with a halfway decent machine at home or at work could hope work with structures up to a similar size without too much trouble.
For many proteins and studies, the authors believe commonly available hardware should be enough to get something useful done without stretching budgets too far. Many computational studies don’t describe their programming or computing environments in much detail, so this one deserves a special mention in this regard. It should be noted the “papers with code” movement is growing, and is vital to reproducibility, and possibly even longevity of an academic’s work.
Roughly, the limit of AF (and AFM) on this computational platform appears to be 3000 residues, and 73/175 (42%) of all complexes are larger than that.
Depending on the speed of computational development, an assembly approach to complex prediction may be needed even for proteins with fewer than 10 chains.
Large multi-chain complexes are still a bit hard for AF2 to predict. According to the authors: “there are 4779 complexes with at least 3 chains, of which only 83 have all chains together in the same PDB entry.” This is a significant number - the group used AF2 on the subcomponents, and then reconstructed them for 175 different proteins using “graph traversal” algorithms. That means they inspect points on a structure similar to a flow chart or network diagram to make calculations.
![Assembly principle for the complex 6ESQ (acetoacetyl-CoA thiolase/HMG-CoA synthase complex)].(https://www.biorxiv.org/content/biorxiv/early/2022/03/12/2022.03.12.484089/F1.large.jpg) Assembly principle for the complex 6ESQ (acetoacetyl-CoA thiolase/HMG-CoA synthase complex). Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/03/12/2022.03.12.484089/F1.large.jpg
Using Alphafold-Multimer or the FoldDock protocol in AlphaFold itself, structures of all pairs of chains were predicted and then larger complexes were assembled from there. They created a new scoring function called “mpDockQ’’ to measure accuracy and combined this with other analyses for each predicted model.
I’d like to once more remark on the effort the authors put into noting software tools and versions used throughout this paper. This makes it much, much easier to reproduce or extend their work. Their code is available here: https://gitlab.com/patrickbryant1/molpc
New 63 knot and other knots in human proteome from AlphaFold predictions3
There’s a little subfield of mathematics known as “knot theory”. It is literally the study of knots - yes, little twists and ties in bits of string.
Some biophysics and informatics researchers use the ideas here to describe and compose strings of DNA, RNA and proteins in particular to understand their structure.
In this study the authors analyzed the human-predicted proteins from AF2 for knots. They identify all 65 knots in the human proteome, including a complex knot called 63 (pronounced “six sub three”). They added them to the KnotProt database: KnotProt 2.0: A database of proteins with knots and slipknots (uw.edu.pl).
Knot theory has a range of applications, but in biology folding of nucleic acids and proteins are two of the most obvious.
They reviewed all the proteins modeled by AF in the human proteome to identify the “dominant knot” in each structure and also the location of the “knot core”. This is the location of the minimal length of peptides required to form the type of knot identified.
The authors raise some broad questions which should illuminate further paths of inquiry if answered:
1) Is the small percentage of knotted proteins observed due to the specificity of the data deposited in the PDB? 2) Are there more complex knots which do not yet appear in the PDB? 3) What is the origin of knotted proteins? 4) Is topology strictly conserved? 5) What role do knots play in proteins?
 Knot 63 in protein form. Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/01/01/2021.12.30.474018/F1.large.jpg
Knot 63 in protein form. Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/01/01/2021.12.30.474018/F1.large.jpg
I’m sure knot theorists must be very happy to see their beloved area of mathematics get such interesting and impactful applications!
Hallucinating Protein Assemblies4
Researchers designed a few proteins with AF2, all ring and loop structures, not found in nature, and also not in the PDB. They developed a system which can invent or “hallucinate” entirely new protein designs. The high level process is straightforward:
- Set the length and number of binding sites of sub-structures to anything you like
- Use them as inputs to a randomised tree search
- Use AF2 for error correction function of the search in (2)
This generates a new random sequence of amino acids with some very interesting and intricate structures. The authors did this many times for many different parameters. They then made some synthetic DNA to encode these protein sequences and inserted them into batches of E.Coli bacteria.
After expression of these proteins in E.Coli, these “hallucinated” designs were evaluated using a range of structural analysis tools. Readers may know of one technique, x-ray crystallography, where proteins are crystallised in a liquid solution and then x-rayed to visualise their structure.
 RoseTTAFold2 predictions aligned to design models of EM-verified hallucinated proteins.. Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/06/09/2022.06.09.493773/F13.large.jpg
RoseTTAFold2 predictions aligned to design models of EM-verified hallucinated proteins.. Sourced: https://www.biorxiv.org/content/biorxiv/early/2022/06/09/2022.06.09.493773/F13.large.jpg
Authors conclude this approach allows the design of novel, complex higher ordered protein structures with less user input and no need for a hierarchical docking approach. It could make for a highly scalable production architecture given the simplicity of both the hallucination and expression process.
Applications could include new nanomaterials or even composite nanomachines, and large proteins with complexity comparable to that found in nature
AlphaFold is no longer alone…
OpenFold OpenFold has recently been released - an open “competitor” and “faithful reproduction” of AF2 with its own code, models and similar results. The OpenFold Consortium has been formed to support open source software tools for biology and medicine. The code is available at https://github.com/aqlaboratory/openfold and is a project of the Open Molecular Software Foundation - https://omsf.io. It is built on PyTorch, and is trainable, supports GPU and most of the code linked above written in the Python programming language.
OmegaFold Another implementation, OmegaFold, appears to have scored significantly higher in LDDT and TM-SCORE on single-sequence data than AF2. The paper and code have now been published, with some diagrams and images released on twitter by the PI, Jian Peng.
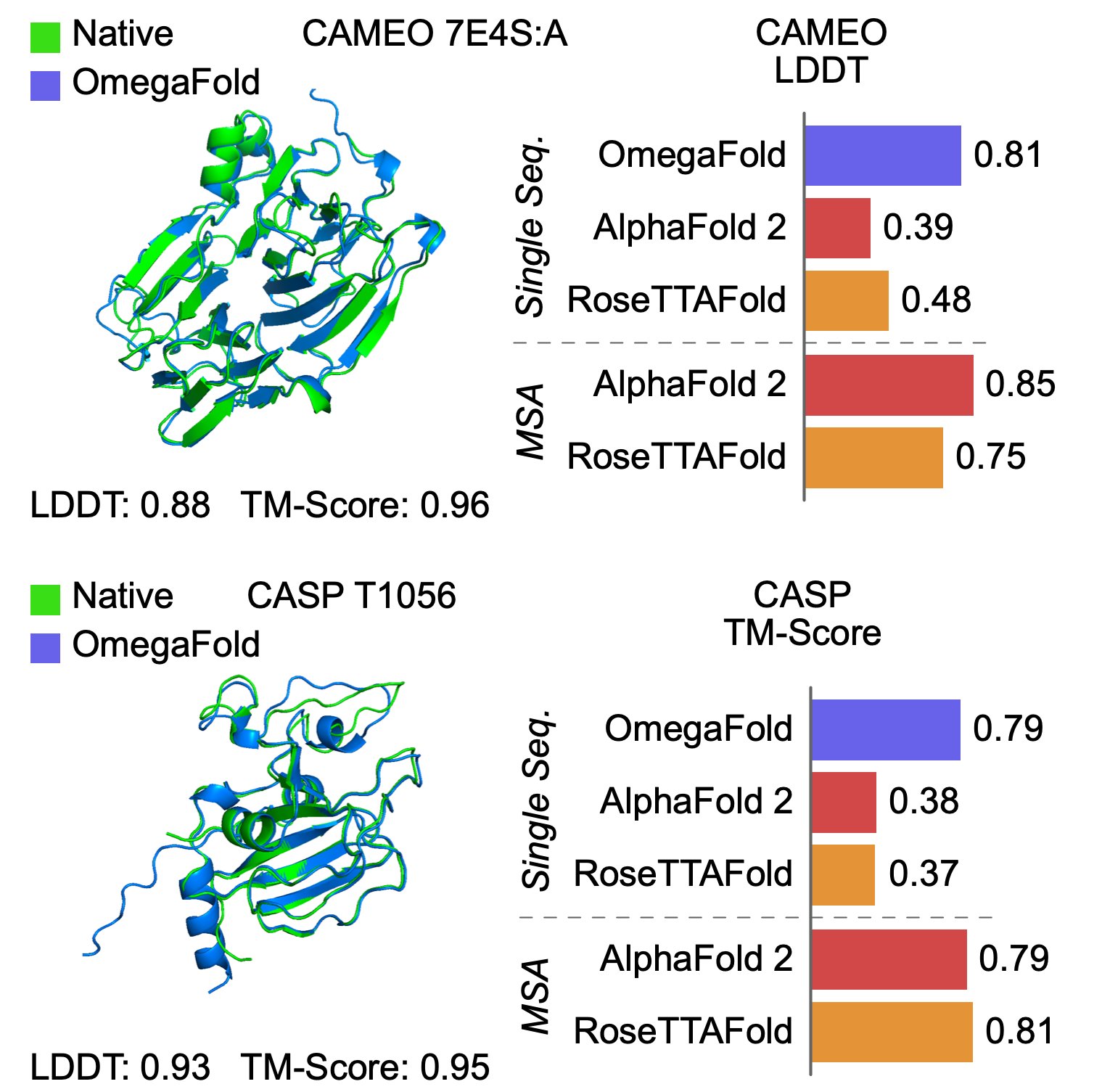
A previous publication from Jian is the “Fast Fold” paper, which significantly increased training speed of AlphaFold 25.
According to Jian, OmegaFold is developed by the Helixon Bio team: https://helixon.com/. Helixon Bio was founded in June 2021, and is raising capital. Based in Beijing, their website: https://www.bioworld.com/articles/520457-helixon-nets-75m-for-ai-drug-development-platform?v=preview
The paper in preprint form on the biorxiv: High-resolution de novo structure prediction from primary sequence | bioRxiv The code for anyone who’d like to inspect and run OmegaFold themselves: GitHub - HeliXonProtein/OmegaFold: OmegaFold Release Code
This is, of course, a short sample of the growing list of AlphaFold applications.
Many of the examples presented here are still in the pre-print stage, so it will be very interesting to see these approaches make their way out into the world and become mature applications. The work for CASP 15 is still ongoing, with modeling and prediction work ending in August and final presentations at the main event in December. At this stage there is talk of a number of entrants outperforming AlphaFold’s previous results at CASP 14.. If all goes to plan, we’ll see the full results at the conference at the end of the year.
###
References
-
https://www.biorxiv.org/content/10.1101/2022.02.02.478886v1?rss=1 AlphaFold accurately predicts distinct conformations based on oligomeric state of a de novo designed protein
-
https://www.biorxiv.org/content/10.1101/2022.03.12.484089v1?rss=1 Predicting the structure of large protein complexes using AlphaFold and sequential assembly
-
https://www.biorxiv.org/content/10.1101/2021.12.30.474018v1?rss=1 New 63 knot and other knots in human proteome from AlphaFold predictions
-
https://www.biorxiv.org/content/10.1101/2022.06.09.493773v1 Hallucinating Protein Assemblies
-
FastFold: Reducing AlphaFold Training Time from 11 Days to 67 Hours https://arxiv.org/abs/2203.00854
-
Highly accurate protein structure prediction with AlphaFold https://www.nature.com/articles/s41586-021-03819-2